Yahoo Finance
Yahoo Finance Coronavirus Vaccine Progress Accelerates
Mentioned: Alphabet Inc (GOOG), Catalent Inc (CTLT), Translate Bio Inc (TBIO), Moderna Inc (MRNA), Adaptive Biotechnologies Corp (ADPT), BioNTech SE (BNTX), Thermo Fisher Scientific Inc (TMO), Emergent BioSolutions Inc (EBS), Incyte Corp (INCY), Hologic Inc (HOLX), LabCorp (LH), Quest (DGX), Apple (AAPL), Regeneron (REGN), Roche (RHHBY), Sanofi (SNY), Novartis (NVS), Lilly (LLY), Gilead (GILD), Pfizer (PFE), Abbott (ABT), Glaxo (GSK)
Editor’s note: This is the second of two updates to Morningstar's COVID-19 forecast. In the first part, we looked at plans for easing the U.S. lockdown. Read the latest on how the coronavirus is rattling the markets and what investors can do to navigate it. |
We think widespread availability of a coronavirus vaccine or curative treatment will probably be necessary to completely remove social distancing measures. In our base-case U.S. scenario, we expect states to wait for reassurance that the COVID-19 spread has flattened before beginning to reopen businesses, and we assume a 30% reduction in visits to nonessential businesses from baseline by the end of the year. This is a significant improvement from levels at the end of March (65% reduction) and should improve further in 2021 with the availability of a coronavirus vaccine. We assume that less than 10% of the U.S. population is infected by the end of 2020, with a 0.7% death rate, as improving levels of testing help control the spread of the disease.
Remdesivir’s recent emergency use authorization in severely ill COVID-19 patients is a turning point, but efficacy to date does not look strong enough on its own to justify relaxing lockdowns. We project $2 billion in peak sales in 2021, assuming an eventual U.S. price (after donated supplies) of around $500 per treatment.
Repurposed drugs could be the next wave of approvals. We expect IL-6 antibodies like Actemra and Kevzara could find a niche among the most severely ill patients by this summer, with data starting to read out on JAK inhibitors like Jakafi and Olumiant this summer, as well.
Novel, targeted antibodies, led by Regeneron’s antibody cocktail, are likely to be effective but are behind this schedule; they could be available late this year for high-risk populations.
While coronavirus vaccines have yet to produce clinical data, encouraging preclinical data, strong partnerships and funding, and rapid clinical progress all seem to indicate that use in high-risk populations could be possible by the end of 2020, with tens of millions of doses potentially available by that point. If just two of these vaccines succeed, we could have enough supply to protect high-risk populations in late 2020 and for broader vaccination as we enter 2021.
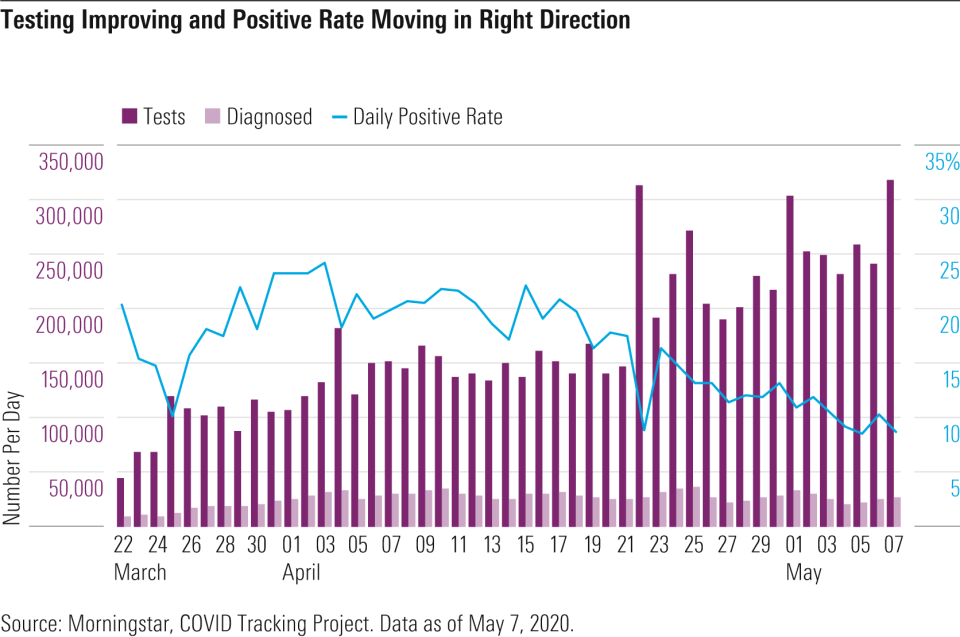
Considering the utter importance of diagnostic testing to help the U.S. begin resuming economic activity in a manner that minimizes the prospect of increases in infection, we remain concerned about national testing capacity meeting necessary levels both in terms of volume and accuracy. The United States was already behind the eight ball when SARS-CoV-2 infections began to rise in February and March. Once the Food and Drug Administration began to approve COVID-19 diagnostic tests under the Emergency Use Act in March, we began to see new tests roll out and capacity for diagnostic testing rise. At the start of May, the U.S. was above 300,000 tests per day, which gets us to roughly 2.1 million tests per week.
Nevertheless, this remains far lower than the 500,000 to more than 1 million tests per day that we see as the midrange of what epidemiologists and economists have projected is required to keep the COVID-19 infection from overwhelming our finite healthcare provider systems.
U.S. testing czar Adm. Brett Giroir says our testing capabilities are in the right ballpark for reopening in May and that four types of testing are needed: surveillance (testing hundreds of thousands a month to get a radar view, even among asymptomatic), symptomatic (millions of tests per month to get a nearly 10% positive rate, meaning 10% of tests coming back positive ), contact tracing (five people per confirmed case on average), and antibody testing. A 10% positive rate is the maximum recommended by the World Health Organization and a level the U.S. recently reached, based on daily data. Cumulative testing results as of May 7, with more than 8 million tests run, show an overall positive rate around 15%.
In addition, comparing U.S. testing rates to other countries gives some perspective. This comparison shows the slow start the U.S. made in ramping testing, and while there is a clear improvement over the past couple of months, the extent of the U.S. outbreak should justify even higher testing rates.
Another way of examining this is from the perspective of positive rates--the percentage of tests coming back positive. While the U.S. is making progress on positive rates, we have just barely reached the 10% threshold as a country, and some states are well above. South Korea was at 2% (roughly 10,000 positive tests out of 600,000), and we are higher than every other developed country outside the United Kingdom, which is at 30%.
Diagnostic Testing and Antibody Testing Serve Two Different Strategies
To open up the economy while minimizing the spread of COVID-19, we need two general types of tests. First, we need diagnostic testing to help identify which patients are in the active phase of infection and, thus, shedding the virus to others. This is the information that is most useful for tamping down the spread of COVID-19 so that infectious patients can be quarantined. Asymptomatic patients could be tested either through surveillance (random sampling) or contact tracing (if disease detectives found the person to be in recent contact with a confirmed patient). Infected patients could be most contagious before becoming symptomatic, making quarantine based on diagnostics rather than symptoms more effective. Dr. Anthony Fauci, director of the National Institute of Allergy and Infectious Diseases, has estimated that 25%-50% of patients are asymptomatic. While some asymptomatic infected people go on to develop symptoms, some do not: 18% of infected patients on the Diamond Princess cruise ship in early February remained asymptomatic. In addition, at least 40% of infected patients were asymptomatic in a broad two-stage survey in the town of Vo’, Italy, and in a random sample by DeCODE in Iceland.
Second, we also need COVID-19 antibody (serology) tests that can provide a better picture of how widely the SARS-CoV-2 virus has spread throughout the larger population, and the proportion of the population that has already cleared the virus from their systems and who may enjoy some lingering immunity to SARS-CoV-2. This test contributes information that helps us more accurately estimate prevalence and how close we might be to reaching herd immunity, generally seen with around 60% of the population.
We think that at this point, the diagnostic test remains the most important testing mechanism, particularly given how low the SARS-CoV-2 infection rate seems to be. We think prevalence is still probably below 5% based on antibody tests among samples in different geographic markets; two studies in California have put prevalence at 2.8% (Santa Clara County) and 4.1% (Los Angeles County), although these studies (and antibody tests in general) have been widely criticized for their potential to produce artificially high prevalence rates, or false positives. One key area of higher prevalence is New York City, which has been hit hardest by COVID-19 and where a recent analysis pegged seroprevalence at 20% (although infection rates in most of the state beyond NYC are in the low to mid-single digits). This still leaves approximately 95% of the U.S. population vulnerable to infection who will need access to diagnostic testing if they are to resume economic activity.
Unfortunately, diagnostic testing is more labor-intensive, slower to turn around results, and reliant on trained professionals to both acquire the specimen and to run the tests in the lab. As a result, diagnostic testing is more challenging to scale up to required levels. Antibody tests are far easier to scale up because they are blood-based, easier to process, and tap into existing infrastructure and labor that can handle significantly higher throughput. Both LabCorp (LH) and Quest (DGX) have extensive networks of patient service centers where specimens can be collected. These companies may even assemble mobile teams of phlebotomists to do blood draws for antibody tests on site at company locations in the second half of the year, boosting access. By the second half, we assume concerns about false positives with the initial tests will fade, particularly given that newer tests, like Roche’s (RHHBY) serology test, have much higher specificity (99.8%) that should reduce false positives regardless of how low the true prevalence of antibodies is in the population. That said, immunity benefits from the antibody are still unclear. Lower or undetectable antibody levels in some recovered patients may not be protective, or may fade in two to three years, although memory B cells (harder to detect than antibodies) could still protect patients.
Antigen tests are just entering the market, and while they provide potential for higher capacity in the long run, we’re concerned about test accuracy and scale (Quidel can provide 200,000 tests per week, scaling to more than 1 million a week in several weeks).
There is emerging research on using saliva samples for RT-PCR diagnostic testing, which suggests it is as good as, if not better than, the nasal-pharyngeal swab that currently dominates those tests. If patient data continue to bear this out, we think shifting to saliva specimens could relieve some of the factors that have slowed diagnostic testing. Experts say that the rush for coronavirus tests has led to low sensitivity, with perhaps a third of patients who are infected getting a negative test result, versus over 90% sensitivity for most PCR-based tests, but saliva-based testing could make it easier for healthcare professionals to get good samples and improve sensitivity of the test.
Aside from the prospect of testing saliva, the current picture of capacity for RT-PCR tests, based on the largest manufacturers, looks far better in theory than in practice. We have examined U.S. capacity by the base of machines, throughput, and cycle time. Based on that, we have estimated total capacity of U.S. existing lab infrastructure can get us close to 5 million diagnostic tests per day. In other words, if we squeezed out every last bit of efficiency on our collective installed base of lab equipment--running full batches, with consecutive cycles, for 16 hours a day, seven days a week (and eliminating all other non-COVID-19 tests that might otherwise be running on that equipment).
In addition to the installed base, U.S. capacity to run tests is limited by the number of tests and kits available. The manufacturing of tests has accelerated but still significantly lags the capacity of the installed base to run them. Nonetheless, our estimate of the number of tests available daily gets us to more than 600,000, which could meet the numbers that public health officials have suggested would be adequate for safely reopening the economy.
However, these estimates reflect theoretical capacity, which is far more hopeful than de facto utilization, which gets us closer to the peak of 315,000 tests in a day. There are any number of bottlenecks and pain points that prevent us from optimizing existing capacity, and many of these inefficiencies reflect the relatively decentralized lab testing environment in the U.S.
Key Obstacles to Optimizing Diagnostic Testing Capacity
Unlike the diagnostics lab industry in other countries, including Australia and the U.K., the U.S. remains a relatively fragmented and decentralized market. Even Quest and LabCorp--the two largest independent reference labs--have only captured an estimated 22% market share. Approximately half the diagnostic market is still controlled by the smaller, hospital-based labs. Considering that most of the competitive labs operate independently and that the federal government has failed to step in with leadership, it’s not surprising that we haven’t seen a coordinated effort to ramp up efficient use of capacity to handle the new COVID-19 tests. This is the overarching theme, and there are additional specific factors that are causing friction in efforts to scale up. There is no magic bullet to be found. Instead, the current situation is more akin to a rowboat that has sprung multiple leaks.
Tests That Are Largely Proprietary to the Equipment
The razor/razor blade model is standard for diagnostic equipment makers, with the proprietary consumables offering a high-margin stream of ongoing revenue once the main machine is installed. For example, Roche’s COVID-19 RT-PCR tests must be run on its cobas 6800 and 8800 high-throughput machines. This means that individual hospitals and integrated delivery networks that are collecting patient specimens with the Roche test must figure out which labs have those Roche cobas machines. This issue is somewhat mitigated if the hospital or network has contracted with LabCorp or Quest to handle some of its lab testing. According to Roche management, there are 122 cobas machines installed in 60 labs across the U.S. LabCorp and Quest are some of Roche’s largest customers, and each has its own information technology systems that provide information on tests coming in and capacity throughout its network of labs. If the hospital hasn’t contracted with this type of larger independent lab network, it may not have much visibility on where there’s capacity to run those Roche tests it has collected, which could lead to longer turnaround times and inefficient use of the installed base.
We suspect this proprietary test-to-equipment framework has been particularly cumbersome for Abbott (ABT), which introduced its COVID-19 diagnostic testing fairly early but had not seen its throughput of COVID-19 tests accelerate to the level expected. We think this is because Abbott’s roughly 175 m2000 machines scattered across the U.S. are in smaller labs. The m2000 is not widely installed at Quest’s network of labs, for example. Molecular diagnostic equipment at Quest is dominated by Roche and Hologic (HOLX). We suspect there may be a similar situation at LabCorp. If most of Abbott’s m2000 machines are not linked to a wider network, it won’t benefit from information flow that can identify and match available machine capacity to incoming tests.
Need for Trained Personnel
This is an issue on two fronts. First, we need trained medical professionals to obtain the specimen via nasal-pharyngeal swab. Capturing that nasal swab can be risky because it puts the medical professional close to the patient. Additionally, swabbing can make patients sneeze, which could further spread the virus if they’re infectious. Second, we also need trained technicians to run the diagnostic machines. These are the people who transfer and prepare the specimens for PCR testing in the lab and monitor quality controls along the way. The dearth of trained lab professionals has been a chronic problem in the industry, even before the COVID-19 pandemic. The prospect of increasing capacity by installing more machines is likely to be constrained by the limited number of people who know how to operate them.
Inadequate Supplies of Personal Protective Equipment for Medical Professionals
The well-documented PPE insufficiency has also hampered the ability of medical professionals to collect specimens from patients. As discussed earlier, the nasal-pharyngeal swab can result in the patient sneezing on the healthcare worker, which means the medical professionals need substantial protective gear to do their jobs safely. If the saliva-based RT-PCR test offers comparable or superior specimen capture, shifting over could relieve some of the pressure on the healthcare workers, in terms of needing less training and PPE.
Inadequate Reagents and Other Supplies for Tests
On a preliminary basis, we have run across reports of healthcare providers running out of reagents, nasal swabs, and transport media supplies necessary to capture and preserve specimens. Test makers have suggested that they are sending out sufficient supplies with their tests. We remain skeptical that supply has matched demand. The bandwidth for producing those enzymes and primers tailored to the SARS-CoV-2 virus remains somewhat limited. These enzymes are biological molecules that are typically produced and then purified on a small scale. Increasing quantity of production will take time.
Restrictive Conditions on Testing for Patients
Severe test constraints in the early stages of the COVID-19 crisis in January and February meant that healthcare providers tried to limit the tests to symptomatic patients, those who were in contact with COVID-19 patients, and first responders. Now that more tests are available, providers can expand their guidelines for testing. However, the Centers for Disease Control and Prevention has not led the way in setting more liberal testing guidelines. Instead, it is leaving it up to the states, which has resulted in a more disjointed shift in making the tests more widely accessible.
Lack of Centralized IT and Logistics Capabilities
We think this, perhaps, is the most significant factor in the near and mid-term that prevents us from turning our theoretical test capacity into reality. Getting the specimen from the healthcare provider to a lab with the appropriate equipment platform that has space in its overnight batches is difficult without a centralized information system. Then, take hundreds of thousands of specimens, multiplied by thousands of healthcare providers, multiplied by hundreds of labs, and by a handful of major equipment platforms. This quickly turns into a massive matching game to get each specimen to the right lab at the right time. White House coronavirus response coordinator Dr. Deborah Birx noted on April 8 that only 10% of the available 1 million Abbott tests for m2000 machines (175 in the U.S.) had been run, probably because the high-throughput machines are not always in locations where they are most needed, and staff and supplies can run short. Federal testing czar Giroir hopes to triple usage to 60% in May.
Both LabCorp and Quest have their own proprietary information systems that can provide insight into equipment platforms and testing capacity on a nightly basis across their own network of labs. However, roughly half the diagnostic industry in the U.S. is housed in hospital-based labs, and they are not looped into such a lab network. Same thing applies to the smaller, regional independent labs. As a result, there’s a fair amount of capacity in our system that is going unused. We think addressing this shortfall will take action on the part of the federal government to centralize information flow related to the incoming tests and individual lab capacity. Based on this administration’s propensity to leave diagnostic testing programs to the states, we speculate that any sort of common information system to unlock capacity in our existing lab infrastructure probably won’t happen unless a different administration takes over next year.
Significantly expanding lab infrastructure will take time. According to the Rockefeller Foundation plan, the U.S. could roughly triple current testing if we efficiently use all labs in U.S. that could do high-throughput testing. According to Rockefeller, it would take about eight months to get to 30 million tests a week, assuming an incredible ramp with strong funding. We could also make more point-of-care tests for rural areas, but building more could run into bottlenecks for China-sourced electronics or reagents. Hologic has been contacted by a range of customers, including departments of public health, hospital systems, and LabCorp and Quest, about buying more Panthers. In the near term, we expect that there will be robust placement of this kind of midsize high-throughput testing equipment. However, production of these machines is not necessarily easy to accelerate three- or four-fold to meet the current demand. Further, it is not clear to us that LabCorp and Quest, as the largest reference labs, are willing to significantly build out and maintain extra capacity that could go unused until another pandemic. Thus, the near- and mid-term expansion of capacity will likely be on the periphery of what we already have. We doubt there will be any tripling or quadrupling of the U.S. installed base of machines.
In the near and mid-term, we expect most of the usable capacity to come from LabCorp and Quest more efficiently using their existing equipment. This will be enhanced by the addition of Hologic’s newest COVID-19 diagnostic testing capabilities, run on its Panther machines, which are located in a range of small and midsize labs.
Point-of-Care and At-Home Diagnostic Testing Unlikely to Be Short-Run Fix
We may see some new innovations to address the testing challenges. Quest management said the current administration has encouraged the firm to think creatively about how to bring on more capacity in different ways. The CARES Act also contains more than $1 billion dedicated to a competition to develop new diagnostics that can get around these pain points that have slowed COVID-19 diagnostic testing so far.
Abbott is ramping up production of its point-of-care diagnostic test, but we still see this topping out at around 2 million tests per month over the summer, translating to 67,000 tests per day. Also, the Abbott machines for POC testing are likely mainly in doctors’ offices, and most of those offices are closed with the machines trapped inside. Abbott management says there are 18,000 ID Now machines in the U.S., but demand for these machines seems unmet, with the mayor of Detroit asking for just five to put in his hospitals. In other words, Abbott’s machines don’t appear to be translating into usable supply. Until ID Now machines are relocated to where most potential COVID-19 patients will be, we’re unlikely to see POC testing significantly increase.
In addition, POC accuracy will probably continue to trail that of the RT-PCR tests done in the reference lab. Reported 15% rates of false negatives for the Abbott POC test should improve peripherally as medical professionals become more familiar with how to run the test and handle the specimens, but that may not be enough to close the gap with lab tests. The other leap forward for the POC tests is if they can run tests on saliva. If there’s greater and more consistent concentration of the virus in saliva, that could improve accuracy. That said, each machine is severely limited in its capacity; one Abbott ID Now machine can process only 64 tests, if run continuously for 16 hours.
The first at-home tests are hampered by the need for a prescription and a $119 out-of-pocket payment. LabCorp expects that in a few weeks it will have enough supply of the Pixel test to branch out from the initial target of first responders, aiming for normalized production of 100,000 tests per week. We think it could be easy to remove the obstacle of needing a prescription, but LabCorp can’t distribute tests without collecting money from someone to cover costs. The process of working with payers to collect the money would take time, as patients would then need to send insurance information to LabCorp for the lab to close the loop with the patient’s payer, collect payment, and send out the test. LabCorp could also contract with employers directly to pay for kits in bulk, which could then be distributed to employees, but we haven’t seen evidence that this is part of its strategy. Regardless, getting at-home tests in the hands of noninsured Americans seems challenging.
Contact Tracing Offers Incremental Improvement in Fighting Outbreak
Contact tracing can help narrow the field of testing to be more precise, but U.S. health departments don’t seem well equipped using traditional methods. The Association of State and Territorial Health Officials recommends 100,000 U.S. contact tracers to get to a 1:1,000 individual/tracer ratio, using a three-tier response of lay contact tracers, disease investigator specialists (professionals) at the state level (currently only 2,200 DIS positions), and healthcare providers or epidemiologists at the CDC (a COVID-19 corps) to support the other two tiers. In Wuhan, China, aggressive contact tracing was done by 1,800 teams of epidemiologists, with at least five members on each team. Scaling up to a level like China’s would require one investigator per 1,200 individuals, or more than 300,000.
To control outbreaks at an R0 (reproduction number, signifying extent of contagion) of 2.5 (estimate for SARS-CoV-2), we would need to trace more than 70% of contacts of those infected, but the outbreak could be controlled with less than 50% successfully traced, if R0 is less than 1.5. We expect this could mean that if we are using additional measures beyond contact tracing to control the disease, such as social distancing or school closures, we may be able to make some headway even without perfect coverage.
A Boston nonprofit, Partners in Health, is hiring 1,000 people for contact tracing, and New York is launching a massive testing and contact tracing program, particularly in hard-hit, low-income communities, involving tens of thousands of tracers (the state previously had fewer than 1,000), and the state has $1.3 billion in federal funding to support the initiative. San Francisco is also boosting its 40-person contact tracer team to at least 150, or one tracer for every 5,553 residents. However, these are small-scale efforts compared with what we would need for a nationwide program.
Technology could make this process easier and faster. Manual contact tracing (by disease detectives) may not be good enough to slow transmission, but instantaneous tracing (notifying exposed people the minute someone is diagnosed) could be, as used in South Korea and Singapore. Apps are being built to help with contact tracing, and proximity tracking could allow people to self-isolate if they get a notification that they have been near someone who is now confirmed to be infected. While apps in Asia have led to the disclosure of personal info, in the West, a system that uses Bluetooth signaling to record proximity could be less intrusive. In the U.S., many projects have been announced, like Covid Watch, CoEpi, and NextTrace, and the ability to piece all of their data together could become an issue unless we rally around one provider. Apple (AAPL) and Google (GOOG) announced that they are working together to facilitate contact tracing using Bluetooth functionality. To satisfy privacy concerns, rather than using GPS, their platform will focus on codes created when users are within proximity for a certain amount of time; if one is infected, the voluntary app (to be designed by healthcare agencies) could automatically warn close contacts that they may have been exposed. While they are first planning to unify their wireless protocols to allow others to build apps across platforms, we expect their own app could be more reliable than third-party apps. With any app, the idea is to limit the number of people who need to be tested in order to maintain containment of the virus. However, epidemiologists estimate that we would need to see 60% uptake of these apps for them to be effective, and so far, uptake is less than 20% for TraceTogether in Singapore and 40% for Iceland’s Rakning C-19.
Another issue is how Americans will react to technology like contact tracing apps. President Donald Trump may be able to sway Republicans to get on board with tracing despite privacy concerns, but preserving freedom is a critical issue. GPS data could work better than Bluetooth technology, for example, as it monitors location (people and surfaces can trigger infection) but is harder to anonymize and harder to use in use in multistory situations. GPS data is helping enforce quarantine of those infected in Taiwan, as well as location-tracking wristbands in Hong Kong and texting proof of being home in Singapore. However, opinions are mixed; a recent survey by a University of Pennsylvania political scientist saw 70% of Republicans and 46% of Democrats strongly opposing the use of cellphones to enforce quarantines, but more than half of Americans back anonymized government smartphone tracking, according to a recent Harris poll.
Even in countries that have successfully used technology for contact tracing, concerns remain. South Korea passed amendments to its Contagious Disease Control and Prevention Act following its 2015 MERS outbreak to allow override of previous privacy laws during a serious infectious disease outbreak, which allowed disclosure of a wide range of personal information to the country’s federal, state, and local governments, including credit card transactions, location, transit history, and medical records. However, criticism of the level of data released to the public could lead to refinement of this strategy in the future, as significant amounts of this data were also released publicly, when aggregated data could have been enough.
Progress With Treatments
Former FDA commissioner Scott Gottlieb sees a treatment as our best hope for returning to normal this year. We now have two regimens with emergency use authorizations, although safety issues with chloroquine (and recent studies casting more doubt on potential efficacy) put the focus squarely on Gilead’s (GILD) remdesivir.
Following strong data released April 29, Gilead received FDA emergency use authorization on May 1. While not at the same threshold as FDA approval, this allows Gilead (with the direction of state governments) to begin distributing remdesivir to severely ill patients at select hospitals in the U.S. Gilead already has 1.5 million doses of remdesivir, enough to treat roughly 300,000 patients, assuming they receive the shorter, five-day dosing regimen that proved similar to the 10-day regimen in the Simple study. Using this dose, Gilead should have enough for 1 million patients by October and 2 million patients by the end of 2020. Gilead has also announced that it plans to license remdesivir to other firms capable of manufacturing it in Europe, Asia, and the developing world through at least 2022.
We now assume that remdesivir can generate $2 billion in sales in 2021, but sales this year will be significantly lower (due to donations and limited supply), and we only model sales extending to 2023 (as we assume progress with a coronavirus vaccine will make stockpiling and demand less common). Our model assumes a price tag of $500 per treatment, a middle ground between the $5 per five-day course expected to help Gilead recoup manufacturing costs and the $4,500 price ceiling estimated by the Institute for Clinical and Economic Review, using a $50,000 per quality-adjusted life year threshold. We believe this will be more than enough to recoup the $1 billion in remdesivir-related development and manufacturing costs Gilead expects to incur in 2020, but not enough to significantly swing Gilead’s valuation.
Remdesivir’s efficacy so far is impressive and looks likely to reduce mortality rates from COVID-19 (although mortality data so far isn’t statistically significant, it shows a trend to a benefit). However, these improvements fall short of what we see as necessary for our former base-case scenario, which assumed that efficacy would be strong enough to allow wider relaxing of lockdown measures on its own.
Several other drug candidates are also moving through testing. Among anti-inflammatories, we expect IL-6 antibodies like Roche’s Actemra and Regeneron (REGN)/Sanofi’s (SNY) Kevzara could find a niche among the most severely ill patients by this summer, although initial data from Kevzara was disappointing (development is now focused on critically ill patients). Data could start to read out on JAK inhibitors like Incyte (INCY)/Novartis’ (NVS) Jakafi and Incyte/Lilly’s (LLY) Olumiant this summer, as well, although we have limited view on their potential efficacy. Targeted antibodies, led by Regeneron’s antibody cocktail, are among the most likely to succeed but are behind this schedule, and we don’t expect them to be available until late this year for high-risk populations. While convalescent serum and hyperimmune globulins from plasma firms could also be quite effective, scale is severely constrained; according to Adaptive Biotechnologies (ADPT), convalescent antibodies can treat only one or two patients for every donation from a recovered patient, and quality can vary.
Future trials will be more likely to focus on combinations with remdesivir, and a trial adding Olumiant to remdesivir (to enroll 1,000 patients) began May 8. Pfizer’s (PFE) protease inhibitor is also entering studies this summer and could be a good candidate for combination therapy with remdesivir.
Coronavirus Vaccine Advancement Even Faster Than Accelerated Timelines
Vaccine development typically takes years--28 years for varicella and FluMist, and 15 years for HPV and rotavirus. We’ve been waiting decades for an effective HIV vaccine, without success. Preparing manufacturing plants to make a new vaccine can cost anywhere from $50 million to $700 million, according to a 2017 paper in the medical journal Vaccine, and the investments are typically made in a stepwise fashion at different scales, based on the available data and probability of eventual approval. However, if a firm is willing to take financial risks, the process can be accelerated.

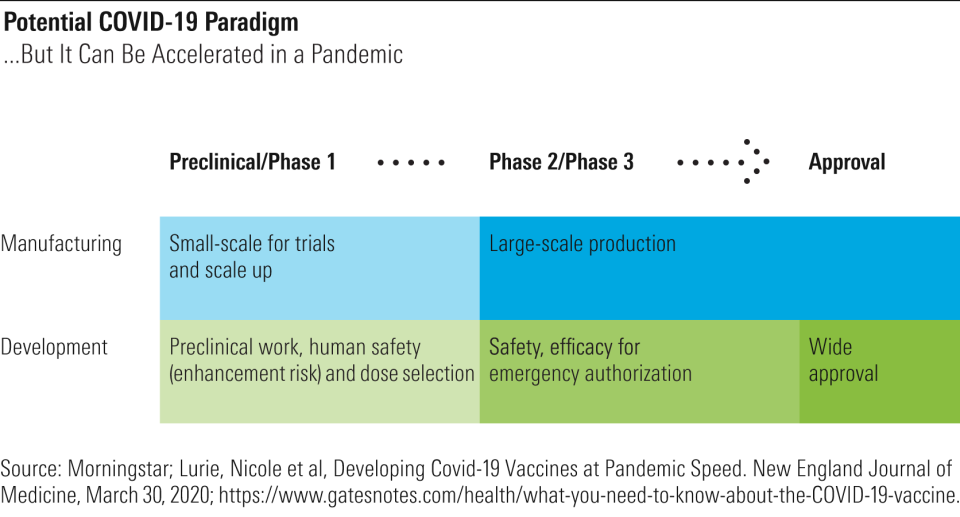
For example, rather than waiting for confirmation of efficacy, a firm can move to large-scale production while efficacy is being tested. Groups like the Bill & Melinda Gates Foundation are taking on some of this risk by offering to pay for manufacturing capacity for a handful of promising vaccines. Government entities (the U.S. Biomedical Advanced Research Development Authority) and public-private alliances (Coalition for Epidemic Preparedness Innovations) are also rapidly offering funding.
Also, rather than building manufacturing capacity, the accelerated process requires partnering with contract manufacturers or large pharma firms to buy extra capacity. This still requires tech transfer for the partner to be able to manufacture the new coronavirus vaccine, but if done early enough, it allows large-scale production to ramp during the second phase of trials. In addition, capital expense is significantly lower for cell-free manufacturing (less than $200 million for nucleic acid vaccines like Moderna’s (MRNA) versus up to $1 billion for more traditional vaccines), which requires smaller reactors and purification tools than recombinant protein vaccines and can be more flexible in switching from one vaccine to another, lowering the risk from investing in manufacturing of any one vaccine in its portfolio. Several of the most advanced programs are nucleic acid (RNA or DNA) vaccines.
Finally, the approval would come in two stages. Broad approval would still be reserved for when a larger phase 3 trial is completed, but the FDA could grant an early, accelerated approval (or emergency use authorization) based on phase 1 and phase 2 safety and efficacy data.
FDA Likely to Accelerate Coronavirus Vaccine Approval if Pandemic Continues This Fall
The FDA’s perspective on approval of a coronavirus vaccine will probably vary depending on how extensively the pandemic is still spreading in the U.S. by the time we have phase 2 data, which will likely be late this summer. With some circulating virus, firms should be able to conduct larger, controlled phase 2/phase 3 studies that can confirm whether antibodies generated in response to a vaccine actually protect individuals from infection. The FDA can also look at antibody levels generated by the vaccine and try to determine whether this could be a biomarker for protection. The second is what Moderna has referred to as looking at “serological correlates” of protection. This can be done by comparing to preclinical models of the vaccine offering protection, or to antibody levels in convalescent serum (from patients who have recovered from infection). If the virus is widespread--and need for the vaccine is most urgent--then Fauci and FDA officials have noted that an emergency use authorization could be made based on phase 1 and animal model safety data as well as efficacy data from phase 2. Such a decision would need to weigh the safety risks; while data through phase 2 should clarify such risks as viral enhancement, it will not be able to isolate any ultrarare risks, which can only be found by testing several thousand volunteers.
If there are a limited number of outbreaks by late this summer, when larger phase 3 studies might be starting (a good problem to have), there are also creative ways of potentially accelerating development to stay ahead of a second wave. The most ethically controversial is challenge studies, which offer a faster option than traditional phase 3 studies by deliberately exposing volunteers to the virus to test a vaccine’s efficacy. For example, 1 Day Sooner is working to qualify healthy, low-risk volunteers for a potential challenge study, with more than 14,000 volunteers listed on the website as of May 8. However, there are serious ethical questions about deliberately infecting patients in such studies, even with their permission. Another way to accelerate development and reduce the number of placebo volunteers is to test multiple vaccines in one trial with one shared control arm; normally, drugmakers avoid this sort of trial, as it forces head-to-head comparisons, but this situation may be exceptional.
We expect initial approval could be reserved for a high-risk population, including the elderly, those with pre-existing conditions, and essential workers (including healthcare workers). We expect that children--who rarely suffer serious consequences from infection and would have the most to lose from theoretical severe but very rare side effects of vaccination--would not be vaccinated until longer-term studies are completed. Vaccination could also be focused on areas with the largest outbreaks if supply is limited.
Probability of Approval Looks Quite High, at Least for a Handful of Coronavirus Vaccines
Recent analysis examining the probability of success of different types of therapeutics, using data from a variety of small- and large-cap drug firms from 2000 to 2015, indicated that vaccines have the highest overall probability of sales, 42% from phase 2 start to approval, and this now stands at 45.8%, using the traditional slow and steady development pathway. In addition, researchers are already familiar with coronaviruses, and particularly familiar with designing vaccines for other serious coronaviruses like SARS and MERS. Key parts of the virus are conserved in SARS-CoV-2 (overall, the viruses are 80% identical), so many vaccines target the spike protein or a portion of it. Researchers see this as unlikely to mutate rapidly, as well, as it is critical for viruses to be able to enter the cell. We think this increases the likelihood of approval. Also, given the sheer number of coronavirus vaccines and the diverse nature of their designs, we would expect some attrition, but some success. Bill Gates recently noted that of the 115 vaccine candidates in the pipeline, 8-10 look “particularly promising,” noting that 60% efficacy would be able to produce herd immunity if we eventually vaccinate roughly 7 billion people, or most of the world’s population. This estimate is likely based on SARS-CoV-2’s reproduction number of 2.5, which means that if 60% are immune and only 40% are vulnerable, the effective transmission rate would decline to 1, meaning flat levels of transmission.
Even if trials complete, plenty of skeptics point to the fact that manufacturing validation typically takes several months, not to mention the manufacturing ramp. However, we think there have been enough partnerships announced to expand manufacturing capacity, as well as parallel programs at several firms, that the capacity should be there if the vaccines are safe and effective, and we assume the FDA would approve the manufacturing facilities at a breakthrough pace. As long as more than one coronavirus vaccine makes it to emergency authorization, that would also relieve some of the pressure on manufacturing to surpass the 1 billion mark. Newer technologies mean much easier scale-up--mRNA vaccines are easier to produce than antigen-based vaccines and require very small doses. DNA and RNA platforms have the most potential for rapid development, followed by recombinant-subunit vaccines.
Virus Enhancement a Key Safety Risk
Until we see evidence that vaccinated patients have been challenged with the virus, we won’t know whether the antibodies created in response to the vaccine paradoxically reduce a patient’s ability to fight a virus. Immune enhancement can be due to antibody-dependent enhancement (antibodies help infection spread) or cell-based enhancement (allergic, type 2 helper T-cell-based inflammation). Allergic inflammation was an issue for at least one SARS vaccine in older mice when they were challenged with the virus. With Sanofi’s Dengvaxia, a vaccine against one of dengue fever’s four serotypes, later infection with a different serotype led to much more severe illness than in those who had not received the vaccine. An RSV vaccine tested in the 1960s caused infections to become much more lethal due to allergic inflammation. Fauci has noted that the key concern in phase 1 is enhancement of the virus, and studies are now in progress for several vaccines.
Leading Coronavirus Vaccine Programs Could Offer Significant Capacity by Year-End
While vaccines have yet to produce clinical data, encouraging preclinical data, strong partnerships and funding, and rapid progress into large phase 1/phase 2 studies all seem to indicate that use in high-risk populations could be possible by the end of the year, with tens or even hundreds of millions of doses available. While we doubt that all vaccines will see strong enough safety and efficacy to reach approval from the FDA, several stand out with a head start and aggressive plans for supplying the market. If just two or three of these vaccines succeed, we could have enough supply to protect high-risk populations in late 2020 (allowing further relaxing of distancing measures) and for broader vaccination as we enter 2021.
Moderna’s mRNA-1273
Moderna’s mRNA-based vaccine is a single antigen vaccine targeting the full-length spike protein. Although the firm has no approved therapies, it has data from more than 1,500 patients studied in various vaccine clinical trials attesting to its safety, and patients have developed neutralizing antibodies to virus antigens for all eight of the viruses that Moderna has vaccinated against in trials. We expect rapid acceleration of manufacturing and development timelines (enabled by at-risk manufacturing expansion, government funding, partnerships, and parallel clinical trials) to allow emergency use by late 2020 and approval for wider distribution in 2021.
Moderna shipped the first vaccine to the National Institute of Allergy and Infectious Diseases 42 days after sequence selection, a record for vaccines, with the first patient dosed as of March 16. This phase 1 trial involves a two-shot series in healthy volunteers, first in 45 adults age 18-55 at three different doses (now fully enrolled), and then at these three doses for 30 older adults (56-70) and 30 elderly adults (71 and older). Phase 1 data is still expected in the second quarter. Moderna received FDA clearance on May 6 to begin a phase 2 immunogenicity study in 600 patients, with 300 adults age 18-55 and 300 age 55 and up receiving two doses of either placebo, 50 ug, or 250 ug doses, so this study will run in parallel with phase 1. Phase 3 timelines have moved up aggressively, and the first phase 3 trial is now expected to begin in early summer, with design still being finalized (although this NIH-partnered study will likely be conducted in the U.S. and will compare natural infection rates and symptoms with patients who have been vaccinated with mRNA-1273 or placebo). We think this coronavirus vaccine could be available for emergency use (healthcare workers) in fall 2020 based on phase 1 and phase 2 data (comparing immunogenicity to what is seen in the plasma of recovered patients), and for broader commercial use in 2021 (based on a potential interim look at phase 3 data in late 2020 and full data in 2021). The Biomedical Advanced Research and Development Authority has agreed to commit up to $483 million to push the vaccine forward to approval, through development and manufacturing support (moving to 24/7 manufacturing).
Moderna is ramping up internal manufacturing to allow millions of doses per month in 2020 and tens of millions per month in 2021, and management’s belief that the 50 ug dose could be selected would help expand the reach of Moderna’s vaccine to more patients (the firm’s Norwood, Massachusetts, facility could supply up to 100 million doses a year at this dosage). An April 30 partnership with Lonza boosts capacity by 10 times, with the goal of producing 1 billion doses a year at the 50 ug expected dose (manufacturing at Lonza suites in the U.S. and Switzerland to begin in July).
Johnson & Johnson
Johnson & Johnson (JNJ) selected an Ad26 vector candidate expressing the SARS-CoV-2 spike protein as of March 30. The firm has steadily accelerated development plans, most recently moving up its estimated phase 1 start from early November to September at the latest, with data available December 2020 and potential launch in emergency use in the first quarter of 2021. J&J has also accelerated supply estimates, from an initial estimate of 300 million doses a year in 8-12 months and ready for emergency use by early 2021 to annual production of 600 million-900 million coronavirus vaccines likely by early 2021 and full supply of 1 billion doses by late 2021. Emergent BioSolutions (EBS) will act as one contract development and manufacturing organization beginning in 2021. Catalent (CTLT) will supply large-scale manufacturing capacity as well, ready for 24/7 manufacturing by January 2021. J&J is aiming for a not-for-profit price around $10.
University of Oxford/Vaccitech/Merck KGaA/AstraZeneca’s ChAdOx1 nCoV-19
This chimp AAV vaccine for the spike protein started a large (1,100 volunteers) phase 1 COV001 study in April 2020, with data expected in May for the first group of volunteers (age 18-55), followed by 55-70 and 70-plus cohorts. This is part of a partnership between Merck KGaA and the Jenner Institute (part of a joint venture with University of Oxford and Pirbright Institute). Oxford now has a 10-liter scale process for the adenovirus vector vaccine. They expect data by autumn 2020 from the phase 2/phase 3 portion of the study (5,000 volunteers expected to start enrolling in May), which could allow emergency approval in September, when the first few million doses could be available. Data from a rhesus macaque study was positive in April (six challenged monkeys stayed healthy). AstraZeneca (AZN) joined as global development and manufacturing lead on April 30, with a cited goal of 100 million doses by the end of 2020. The collaboration is not-for-profit during the pandemic.
Sanofi/Glaxo
Sanofi is working with BARDA on a coronavirus vaccine, using proven influenza FluBlok technology (with Glaxo’s adjuvant booster). Sanofi partnered this program with Glaxo (GSK) on April 14 and plans to move a coronavirus vaccine to development in the fourth quarter of 2020, with earliest approval in the second half of 2021. They expect to be able to manufacture a billion doses by April 2021 by sharing their technology (Sanofi’s S-protein antigen using recombinant DNA technology and Glaxo’s adjuvant technology, which can lower the required dose). Sanofi also entered a collaboration with Translate Bio (TBIO) for its mRNA vaccine, which, like the more traditional Sanofi/Glaxo vaccine, could be in a clinical trial by the end of 2020 and be approved by the end of 2021. Translate’s vaccine has been manufactured at a 100-gram scale, and CMO manufacturing of two 250-gram batches per month could add to supply; at Moderna’s midlevel dose, this could vaccinate 3 million people per month initially, but hundreds of millions by the end of 2021 with Sanofi’s help (at kilogram-size batches). Sanofi expects 90 million-360 million doses by the first quarter of 2021.
CureVac
Working with CEPI, CureVac has two mRNA candidates that do not target the spike protein poised to enter trials by summer. CureVac’s mRNA is self-amplifying, which reduces the amount of vaccine needed for manufacturing. While the three approved manufacturing suites could make 10 million doses per campaign, a fourth suite would push this north of a billion per production run. CureVac received funding from the European Commission in March.
Pfizer/BioNTech
BioNTech (BNTX) started a trial in Germany, with the first cohort of 12 volunteers in Germany dosed by April 29, and a U.S. trial (a phase 1/phase 2 trial to enroll 360 adults and elderly volunteers) kicked off on May 5. Fosun has China rights, and BioNTech signed a letter of intent March 17 with Pfizer elsewhere (expanding their flu vaccine partnership to coronavirus). As of early April, Pfizer said it could have tens of millions of doses by the end of 2020 and hundreds of millions in 2021. The first BNT162 study in Germany will test doses between 1ug and 100 ug in 200 volunteers (ages 18-55) for four vaccines (modified mRNA, uRNA, and self-amplifying RNA). SaRNA could have a big dosing advantage, potentially requiring just one dose (others could require prime and boost) and 60 times lower dose versus uRNA vaccine (and perhaps against Moderna’s mRNA vaccine). Pfizer plans to choose the one or two most promising to move forward by June, with the help of data from primates and plasma of vaccinated humans due by late May/early June. Pfizer is aiming for an EUA or accelerated approval in October (with human study and surrogate endpoint), at the start of flu season and a time when the next wave could be building. Pfizer’s Mikael Dolsten said on a call with Stat news that it could focus on the high-risk population, which could be 30%-40% of the broader population.
Fading Antibody Levels and Mutation Rates Unlikely to Derail Near-Term Pipelines
With a new virus, it’s difficult to say how long immunity will last after infection or after treatment with targeted antibodies or a vaccine. We don’t know what antibody level is sufficient to protect a patient, although research on convalescent serum (from recovered patients) is ongoing. For older coronaviruses that trigger common colds, reinfection can occur. However, even if protection is incomplete, experts say antibodies are likely to at least lead to milder infections, and recent research seems to indicate that almost all infected patients produce antibodies. We will very likely need to continue to make new versions of antibodies or vaccines if the virus recurs. However, we think it’s very likely that coronavirus vaccines in development today, if effective in trials, will provide enough immunity to get us through the pandemic.
While SARS-CoV-2 could mutate to become resistant to vaccines currently in development (targeting the spike protein), we think the evidence so far indicates that this is unlikely. RNA viruses--which include viruses like HIV, influenza, and coronaviruses--generally have the highest mutation rates among microorganisms, creating concerns about how long a potential treatment or vaccine could be effective before SARS-CoV-2 is able to mutate around it. HIV vaccines have been in development for decades without positive results because HIV’s surface molecules (envelope glycoproteins) mutate so rapidly, even within one patient, that they can learn to evade treatment and vaccines, and we now have multiple versions of the virus circulating in society. For influenza, we have vaccines, but these are only at best 60% effective in preventing the flu. This is because there are multiple strains of flu with different surface markers, or antigens, and with so many different strains circulating, it is difficult to predict which one will become most dangerous in a given season (and only so many can be selected for the vaccine). In addition, the flu can mutate, creating new versions that would require new vaccines. Flu vaccine effectiveness can be as low as 20%-30% if vaccine manufacturers do not select the viral strain that ends up being most common that season.
While most RNA viruses have an RNA polymerase that operates without proofreading capabilities--meaning that the viral genetic material mutates rapidly--coronaviruses have such large genomes that they do have proofreading, so their mutation rate is significantly lower. In addition, typically, the most crucial parts of the virus--for example, the spike protein (which allows the virus to infect a cell and is the target of vaccines and antibodies), the polymerase (that copies coronavirus genes, targeted by Gilead’s remdesivir), and the protease (that cut peptides to help form key viral enzymes, targeted by Pfizer’s antiviral)--are not subject to as much variation because of their essential nature. While antivirals like remdesivir could pressure the virus and trigger mutations in the viral polymerase that allow higher mutation rates, changing a viruses’ established mutation rate is more likely to destabilize it and weaken it. A recent hypothesis regarding SARS-CoV-2 mutations and increased transmissibility of a new mutant version (Spike D614G) has been released (a preprint article, not a peer-reviewed publication), but Moderna doesn’t expect mutations seen so far to interfere with the efficacy of its vaccine. In addition, if Moderna validates its technology with an initial vaccine, time to availability would be quite short for a new vaccine, with manufacturing capacity already expanded and a much-abbreviated development timeline (like annual flu vaccine changes).
Finally, drug developers are working on ways to make their products less vulnerable to mutations. Antibody developers like Regeneron are also working on potential cocktails of antibodies to make sure that a potential point mutation doesn’t erode efficacy, and Regeneron says that its cocktails work on all of the spike protein variants it’s tested in vitro (Amgen (AMGN) and Adaptive are hoping for two or three in their cocktail, as well). By the time such point mutations develop (if they do), we should have well-developed infrastructure for producing novel vaccines, allowing for a much faster response. In addition, current antivirals could offer some protection despite mutations that lower coronavirus vaccine efficacy.
In Our Pharma and Biotech Coverage, Roche, Pfizer, and Sanofi Undervalued
Gilead’s remdesivir received emergency use authorization from the FDA on May 1 based on strong data from two clinical studies. Although the initial supply will be donated, we expect a $500 U.S. price tag to allow for $2 billion in sales in 2021. The arrival of coronavirus vaccines is likely to make any profits short term, limiting the upside to Gilead’s valuation, and the shares look fairly valued.
Roche is a leading supplier of SARS-CoV-2 diagnostics and antibody tests, which provides some counterbalance to the decline in routine diagnostic tests due to the pandemic. Immunology drug Actemra could also prove helpful in severely ill patients (data should be available this month). While Roche shares have performed strongly during the downturn, they remain slightly undervalued.
Regeneron and Amgen are both racing to bring targeted antibodies to market, which could serve as treatments or prophylaxis for COVID-19. We think Regeneron is a few months ahead, and greater visibility gives us confidence to include $2 billion in potential sales in our model in 2021, similar to the sales potential of remdesivir. However, without clinical data or guidance on pricing, it’s difficult for this to move the meter substantially on our Regeneron valuation, and the shares look slightly overvalued. Amgen’s partnership with Adaptive is encouraging, but we’re awaiting more details before including this in our model, and Amgen shares look fairly valued.
Big Pharma firms like J&J, Sanofi, Glaxo, Astra, and Pfizer are all rapidly accelerating development and manufacturing timelines to bring a coronavirus vaccine to market. Shares for all these firms have held up well during the pandemic, but we think the shares of Pfizer and Sanofi still look slightly undervalued, and Pfizer’s partnership with BioNTech offers several shots on goal. That said, pandemic sales for most of these coronavirus vaccines will likely be not-for-profit.
Most Diagnostics Names Appear Fairly Valued or Overvalued
We modestly lowered our fair value estimates for LabCorp and Quest to reflect loss of revenue from non-COVID-19 tests, which will only be partially offset by COVID-19 tests. If the normal volume of surgeries and elective procedures returns sooner than expected, this could offer upside to our valuations. We think LabCorp’s higher proportion of esoteric tests (hit less severely than routine tests) could allow it to fare marginally better, and shares look slightly undervalued.
Abbott remains overvalued, from our perspective, after we incorporated declines in medical devices to reflect procedures crowded out by COVID-19 through the second and third quarters, as well as softness in nutritionals thanks to global recessionary conditions. Traditional and point-of-care COVID-19 tests support strong molecular diagnostics growth but don’t significantly sway our valuation.
Hologic shares are fairly valued, in our opinion, after incorporating weakness in the women’s health and surgical segments, which is partially offset by COVID-19-driven growth in molecular diagnostics. Considering Hologic’s extensive installed base of Panthers in the U.S. and their presence in the largest reference labs, increased production of SARS-CoV-2 tests could add upside to our valuation.
While Thermo Fisher (TMO) stands to benefit from COVID-19 as one of the developers of diagnostic testing with scale, a pullback in capital spending will modestly affect its instrumentation business. However, Thermo Fisher’s business is less exposed to higher-ticket purchases than many of its analytical instrument peers (and relative to its own business during the last economic pullback), and its strong consumable revenue stream should help the company weather this cycle rather unscathed. The market seems to agree, with the shares trading above prepandemic levels and ahead of our valuation.
This article was written by Karen Anderson, CFA© and Healthcare Strategist at Morningstar, a research partner for Yahoo Finance Premium.
Karen and Preston Caldwell, Equity Analyst at Morningstar, discussed the long-term economic impact of the global shutdown in a special Yahoo Finance Premium webinar on Tuesday, May 19 at 12pm ET. Watch the recording here.
Karen Andersen has a position in the following security mentioned above: DGX. Find out about Morningstar’s editorial policies.
Additional Morningstar research is available in Yahoo Finance Premium. Start your free trial today.*